
Recent Research Highlights
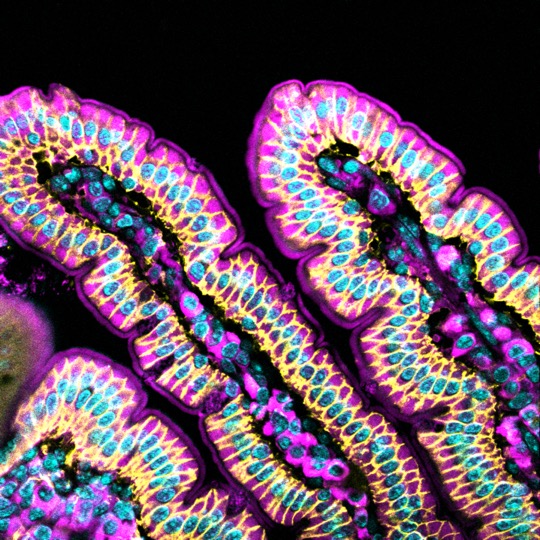
Nature Communications 2020
The ABCG2Q141K hyperuricemia and gout associated variant illuminates the physiology of human urate excretion.
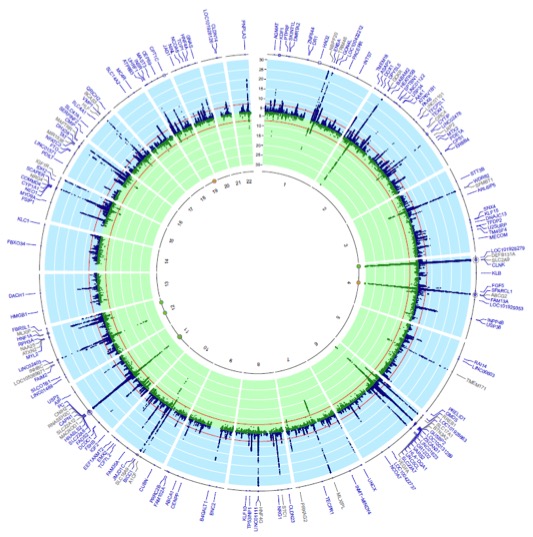
Nature Genetics 2019
Target genes, variants, tissues and transcriptional pathways influencing human serum urate.
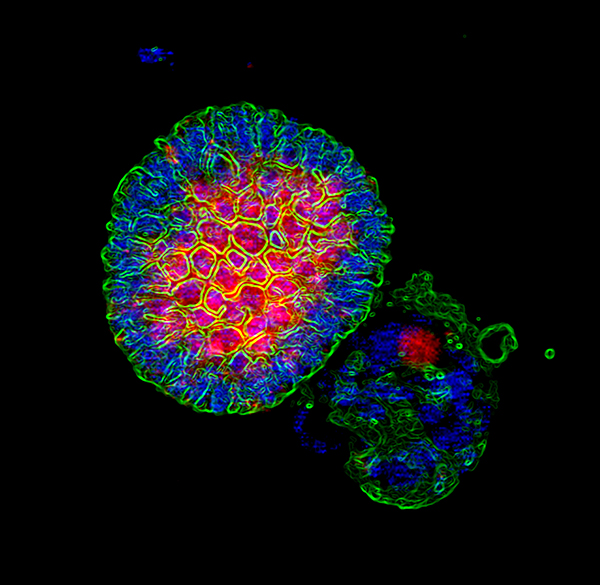
Journal of Cell Science 2020
GDNF drives rapid tubule morphogenesis in a novel 3D in vitro model for ADPKD
Current lab projects
Urate and ABCG2
Urate (uric acid) handling and secretion in humans and the great apes is unique among mammals; we have lost the function of the urate oxidase (uricase) enzyme, the enzyme responsible for metabolizing urate into allantoin. The loss of uricase appears to be adaptive in humans, however it puts humans at risk for retaining too much urate (hyperuricemia), which can lead to gout, kidney disease, hypertension, metabolic disorders and cardiovascular disease. Yet, until recently the transporters responsible for urate secretion and absorption remained mostly unknown.
We discovered that ABCG2 is a novel urate transporter, perhaps the most important secretion mechanism for uric acid in humans, and identified a loss of function mutation that causes hyperuricemia and gout. Importantly the mutation is common, carried by almost a billion people, putting them at increased risk for hyperuricemia, gout, and possibly hypertension and other metabolic diseases.
We use an array of methodologies to better understand urate homeostasis and the role of urate in human disease. Our projects focus on:
- Regulation of urate transporters in the kidney and gut.
- The mechanisms underlying the dramatic differences in how males and females handle urate.
- Understanding how specific human variants in the “big three” urate transporters, ABCG2, SLC2A9, and URAT1 alter function and contribute to disease risk.
- Understanding how hyperuricemia alters metabolism and increases risk of DKD.
- Understanding why urate is an incredibly important risk factor in the progression of chronic kidney disease.
Our urate transporter research is funded by the NIDDK.

Polycystic Kidney Disease
Inheritance of polycystic kidney disease genes causes slow growing kidney cysts with severe consequences for kidney function. Study of disease causation is often obscured by the later stages of a multistage disease process. Our lab focuses on the first stage of ADPKD, cystogenesis, and on the first protein changes that occur upon acute loss of the PKD2 disease gene and protein product PC2. We use a new ex-vivo 3D culture method to grow epithelial kidney tubes to investigate what happens to the nephrons as they transform into cysts with the loss of PKD2. Discovery of the initial steps of cystogenesis after PKD2 loss may illuminate precise drugable targets for the development of future PKD therapeutics.
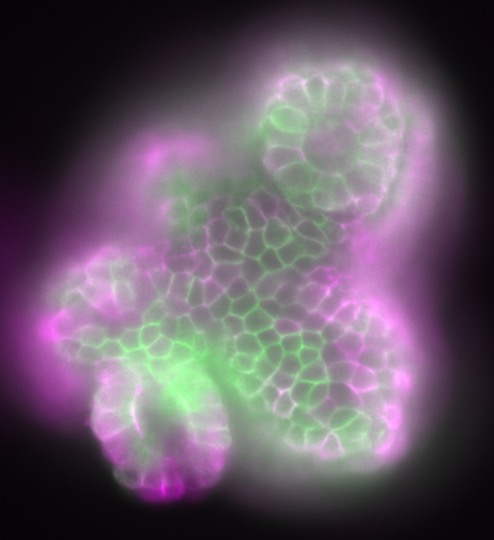
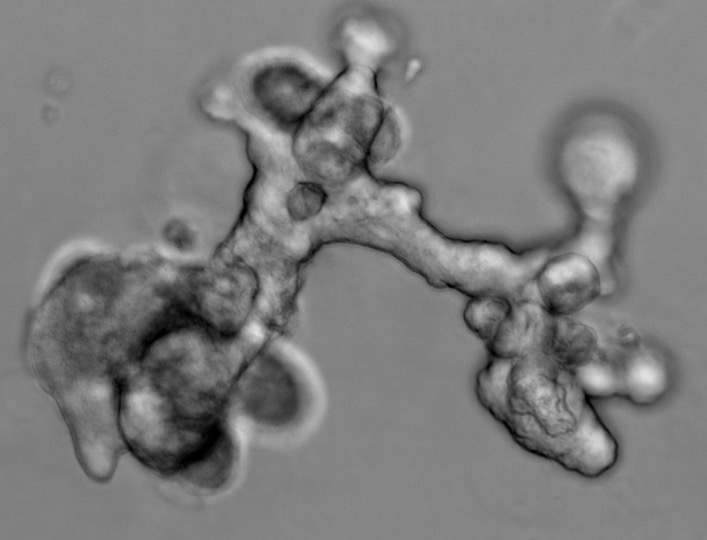
We are also part of the Baltimore Polycystic Kidney Disease (PKD) Research and Clinical Core Center and host the Cell Culture and Engineering Core and provide a number of services for the broader PKD research community.

Our PKD research is supported by the PKD Foundation and the NIDDK.

